Les recherches sur l’histoire de l’humanité ont scientifiquement débuté avec la publication, de 1749 à 1789, de l’Histoire naturelle en 36 volumes de Georges Louis Leclerc, comte de Buffon[1]. À la suite de Buffon, divers auteurs propagent l’idée de l’ancienneté du genre humain et Jacques Boucher de Perthes (1788-1868) lance la science de la préhistoire. La première découverte d’un homme fossile a eu lieu en 1856 dans la vallée de Neander aux environs de Düsseldorf. Cet homme appartient à une entité fossile, celle de Neandertal (Homo neanderthalensis) ayant vécu en Eurasie de -450 000 ans à -35 000 ans environ. Depuis, les découvertes se sont succédé dans le monde entier et ont permis d’établir par approximations successives une phylogénie possible du genre humain. Les récents travaux de génétique des populations et de génomique ont apporté une dimension supplémentaire et continuent d’apporter des éléments nouveaux. L’objectif de cet article est de faire le point sur les dernières découvertes et les hypothèses en cours.
Les connaissances sur l’évolution humaine avant l’avènement de la génétique des populations et de la génomique
Il y a environ -1,8 à -2 millions, ou plus, des populations humaines regroupées sous le vocable Homo erectus ont commencé à se différencier en Afrique subsaharienne[2]. De là, aux environs de 1 million d’années ou plus, elles se sont disséminées vers l’Asie et vers l’Europe de l’Ouest où elles ont donné naissance à l’homme de Heidelberg[3] ou encore à l’homme de Tautavel[4]. Ces hommes datant de la fin du Paléolithique archaïque (-3,3 millions à -1,76 million d’années) ou du Paléolithique inférieur (-1,76 million à -350 000 ans) étaient de petite taille, entre 155 et 165 cm, pour 55 à 65 kg, avec un volume crânien de 800 à 1150 cm3 pour les populations les plus anciennes, 1150 cm3 pour l’homme de Tautavel et 1200 pour l’homme de Heidelberg. Leur visage était caractérisé par un prognathisme important avec un bourrelet sus-orbitaire et un chignon occipital. Ces hommes étaient des chasseurs-cueilleurs, maîtrisant parfois ponctuellement le feu et dotés d’outils de type galets aménagés. L’étude des outils montre une évolution très progressive. Les galets aménagés vont être plus travaillés et à la fin de Paléolithique inférieur se transformer en biface, c’est-à-dire en galets travaillés par percussion sur l’ensemble de leur surface.
À la fin du Paléolithique inférieur ou au début du Paléolithique moyen ou Moustérien (-350 000 à -45 000 ans), ont émergé en Europe les hommes de Neandertal ou Homo neanderthalensis. En quoi L’homme de Neandertal diffère-t-il de l’Homo erectus ? Il est plus grand, plus lourd, plus massif, plus puissant. Il a été capable de s’adapter aux glaciations successives. Sa capacité crânienne atteint 1500 cm3, soit plus que celle de l’homme moderne. D’où viennent ces hommes de Neandertal ? Le plus vraisemblable est qu’ils sont les descendants des Homo erectus qui les ont précédés en Europe. Après avoir évolué en Europe, des populations de Néandertaliens auraient migré vers le Proche-Orient, puis l’Asie centrale, la Sibérie et peut-être même la Chine. L’homme de Neandertal serait donc un Européen descendant des Homo erectus arrivés d’Afrique entre -800 000 et -1 million d’années. Certains qualifient même les hommes du Paléolithique inférieur, au moins ceux d’il y a 4 à 500 000 ans, de Pré-néanderthaliens.
Les outils du Paléolithique moyen sont encore essentiellement des bifaces, mais plus travaillés que ceux du Paléolithique inférieur. Apparaissent aussi des racloirs et des pointes de flèches. Les derniers hommes du Paléolithique inférieur et surtout les Néandertaliens maîtrisent parfaitement le feu. Les premiers foyers aménagés datent en effet de -400 000 ans[5].
En Afrique subsaharienne, l’entité Homo erectus évolue comme en Europe vers une autre entité qui pourrait être Homo rhodesiensis. Entre -150 000 et -200 000 ans apparaît an Afrique subsaharienne un homme plus gracile, plus apte à la course que les hommes de Neandertal ou les hommes de Rhodésie. Il possède la capacité de s’adapter à tous les milieux grâce à ses facultés d’invention et ses capacités de chasse (invention des propulseurs permettant d’atteindre des cibles à grande distance). Cet homme moderne, dit Homo sapiens, va conquérir le monde et atteindre la Terre de feu en passant par le détroit de Behring après avoir colonisé la Chine et la Sibérie. Il arrive en Europe de l’Ouest en provenance du Moyen-Orient vers -50 à -60 000 ans. Il y côtoie les hommes de Neandertal qui vont disparaître vers -30 000 à -35 000 ans. Les causes de cette disparition font toujours débat, mais commencent à être perçues sous un jour nouveau à la suite des récents travaux de génétique des populations et de génomique.
Éléments de génétique des populations
La génétique des populations est une science jeune, créée entre 1920 et 1940. Elle étudie la distribution et l’évolution des allèles au sein de communautés d’individus. Le terme individu vient du latin individuum c’est-à-dire qui est indivisible[6]. Un individu est original. Il n’existe pas deux individus totalement identiques sauf si ce sont des clones ou des jumeaux. Chez un individu tous les éléments sont interdépendants et coopèrent à la vie de l’ensemble. Un individu est caractérisé par un phénotype et un génotype.
Le phénotype est l’ensemble des caractéristiques résultant de l’expression des gènes et de leurs éventuelles interactions avec l’environnement. Deux clones peuvent être morphologiquement différents, bien que génétiquement identiques.
Le génotype est l’ensemble de l’information génétique présent dans chaque cellule. Chaque cellule d’un individu possède la même information génétique que la cellule initiale[7]. Deux clones ont exactement le même génotype. Un génotype est unique et ne peut se retrouver que chez des jumeaux vrais ou chez des clones. Un génotype est caractérisé par l’ensemble du génome[8].
Une population naturelle est constituée d’individus occupant un espace défini à un instant déterminé et échangeant des allèles de manière homogène et régulière par reproduction sexuée. C’est ce qui est appelé panmixie. Les individus de cette population, qui sont tous différents par leur génotype, ont cependant tous les mêmes loci, c’est-à-dire les mêmes localisations de gènes sur les chromosomes. Mais à chaque locus ils ont deux allèles différents, l’un provenant de la mère, l’autre du père. Un allèle est un variant d’un gène occupant une place précise ou locus sur le chromosome. Il peut être dominant ou récessif[9]. Il existe par conséquent une variabilité génétique plus ou moins importante au sein d’une population. Cette diversité est d’autant plus grande que le nombre d’allèles par gène est grand ; c’est ce qui est appelé polymorphisme. Dans une population homogène où règne la panmixie[10], les fréquences de chaque allèle sont proches, c’est-à-dire qu’il n’y a pas d’allèle fortement majoritaire.
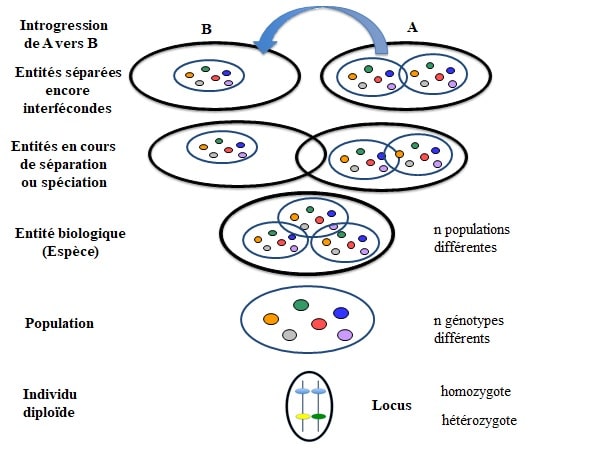
Figure 1. Représentation schématique d’une population, d’une espèce composée de populations différentes à individus diploïdes avec début de différentiation en deux espèces (spéciation) et introgression entre deux espèces non encore séparées génétiquement. Un locus est homozygote quand les deux allèles sont identiques et hétérozygote lorsque les deux allèles sont différents.
Une espèce ou unité systématique de base, a été définie biologiquement par Mayr (1942) comme un groupe de populations naturelles réellement ou potentiellement interfécondes, isolé de tout autre groupement analogue. Les populations constituant une unité de base ont tendance à diverger. En effet, les génomes ne sont pas stables et des modifications se produisent sans cesse au moment de la reproduction sexuée sous l’effet de facteurs internes ou externes. À l’intérieur d’une population naturelle soumise à la panmixie, les échanges génétiques fréquents brassent le tout et homogénéisent les différences internes. Mais les flux géniques peuvent être modifiés par différents mécanismes. La divergence entre population peut alors devenir telles que la reproduction sexuée devienne impossible. Il y a spéciation, c’est-à-dire création d’une nouvelle espèce ou entité biologique de base (Figure 1). L’événement capital dans l’apparition d’une nouvelle entité est la mise en place d’un isolement reproductif. Cet isolement est la résultante de l’apparition de barrières aux flux géniques. Ces barrières peuvent être d’ordre physique, par exemple les espèces occupent des habitats différents : elles peuvent être mécaniques (incompatibilité anatomique des organes génitaux), ou éthologique (attraction sexuelle faible), etc.
Le mode de spéciation le plus courant admis est le mode allopatrique (Turelli et al., 2001). Il est la conséquence d’une fragmentation de l’aire géographique ancestrale de l’espèce au cours du temps. L’émergence d’une barrière physique à la migration permet aux populations d’évoluer séparément du fait de la dérive génétique. D’autres modes de spéciation, dits sympatriques, peuvent se produire au sein d’une population par suite de modifications de l’habitat ou du comportement ou par suite d’autres facteurs (Johannesson, 2001).
En dehors du concept biologique de Mayr, l’espèce peut être appréhendée de différentes manières, les deux principales étant par la morphologie ou la phylogénie.
Le concept morphologique est le plus utilisé. Il consiste à identifier une espèce par les caractères apparents des individus qui la composent (taille, couleur, etc.). La grande force du concept morphologique est d’être applicable partout. Sa faiblesse est de multiplier les taxons ou inversement de regrouper plusieurs espèces non identifiables génétiquement.
Le concept phylogénétique de l’espèce repose sur l’identité génétique et inclut la notion de descendance à partir d’ancêtres communs[11]. Suivant ce principe, une espèce est un ensemble de populations descendant du même ancêtre, maintenant son identité vis à vis des autres espèces et possédant des tendances évolutives et une histoire propre (Wiley, 1978). Ce concept était peu opérationnel jusqu’à l’avènement de la biologie moléculaire et la constitution des banques de données moléculaires. Mais souvent la question est la même que pour la méthode morphologique. Où doit-on placer la limite de l’espèce ?
Dans cet article consacré à l’homme, nous n’utiliserons pas la notion d’espèce. Nous nous contenterons d’utiliser les termes de population ou d’entité biologique. En effet, en anthropologie et encore plus en paléontologie, la notion d’espèce est particulièrement floue. Pour les hommes fossiles nous n’avions, jusqu’à quelques décennies, que des données morphologiques fragmentaires. De plus le genre Homo a une particularité qui lui est propre. Les populations qui le composent ont une propension à migrer massivement et à s’adapter à toutes les conditions de milieu par mise en œuvre de techniques adaptées à toutes les situations. La pression de sélection du milieu en est d’autant diminuée, ce qui limite la spéciation.
Les apports de la génomique
Le séquençage de génomes entiers d’espèces actuelles a commencé en 1995 avec celui d’une bactérie Hemophilus influenzae comprenant 1740 gènes, puis s’est poursuivi en 1998 avec celui d’un champignon, la levure ou Saccharomyces cerevisiae, comprenant 6000 gènes. En 2001, a été publiée une première ébauche du génome humain, suivie de versions plus complètes[12] avec environ 26 000 gènes[13]. Depuis, le séquençage du génome humain se poursuit sur des milliers d’individus, ce qui permet de commencer à définir les diverses populations existant dans le monde. (Figure 2). On peut ainsi déterminer la fréquence relative des différents allèles et reconstituer l’histoire de ces populations.
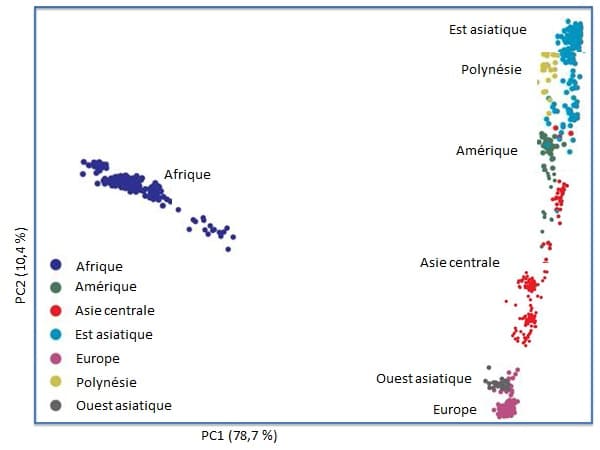
Figure 2. Analyse en composante principale du génome de 850 individus caractérisés par 906 000 SNPs (Single Nucleotide Polymorphism)[14]. Chaque individu est représenté par un point dont la couleur correspond à sa région d’origine. Le pourcentage de variance expliqué par chaque composante (PC1 et PC2) est indiqué sur les axes. D’après Xing et al., Genomics, 2010.
La singularité des populations africaines en termes de distance génétique et de variabilité s’oppose à celle des autres continents (Figure 2). Comme nous l’avons vu au précédent chapitre, la paléontologie classique basée sur les fossiles nous a appris depuis quelques décennies que l’homme moderne, Homo sapiens, est né en Afrique subsaharienne en évoluant à partir d’Homo erectus ou de ses descendants et qu’il s’est ensuite répandu sur les autres continents après la seconde phase dite out of Africa, il y a environ 50 à 60 000 ans. Cet homme moderne a continué à se diversifier en Afrique après cette sortie, mais beaucoup moins que les hommes qui avaient quitté ce continent[15]. Comment expliquer une telle distance entre l’Afrique et le reste du monde et comment expliquer une telle diversité entre les populations autres que subsahariennes ? Les migrations ont évidemment joué un rôle dans cette diversification hors d’Afrique, mais ne semblent pas pouvoir expliquer complètement de tels écarts. La variation génétique humaine actuelle ne semble pas pouvoir être attribuée uniquement à la population africaine ancestrale. Elle pourrait s’expliquer par des apports extérieurs importants, hypothèse qui va être confortée par l’analyse des génomes fossiles.
L’ADN d’échantillons anciens est fragmenté et pollué par de l’ADN de microorganismes divers. Il est donc difficile à extraire et à séquencer et à déchiffrer. Ce n’est que depuis quelques années qu’il est possible d’obtenir des séquences lisibles d’hommes fossiles. En novembre 2006, les premières séquences d’un homme de Neandertal âgé de 38 000 ans et provenant du site de Vindija en Croatie ont été publiées dans Nature (Green et al., 2006). Il s’agissait d’ADN mitochondrial transmis par voie maternelle et d’ADN nucléaire transmis par les deux parents. Selon ces séquences, la date de divergence entre l’homme moderne et les Néandertaliens se situerait entre 461 000 et 825 000 ans, ce qui est assez en accord avec les données paléontologiques. Depuis, de multiples séquences d’ADN néandertaliens ont été obtenues, ce qui permet d’éclaircir en partie notre histoire évolutive.
Une première étude de 2006 avait déjà suggéré qu’un pourcentage non négligeable du génome humain avait des dates de coalescence plus anciennes que ce qui pouvait être expliqué par une seule population ancestrale africaine et avait invoqué de multiples événements de mélanges anciens, dont un provenant des Néandertaliens (Plagnol et Wall, 2006). Il est ensuite apparu que tous les génomes actuels issus de la population qui avait quitté l’Afrique subsaharienne, c’est-à-dire les génomes eurasiens, contenaient des séquences néandertaliennes, alors que les génomes africains n’en contenaient pas[16]. Cette présence de séquences néandertaliennes chez les Eurasiens s’explique par les diverses introgressions[17] qui se sont produites après l’arrivée des hommes modernes au Moyen-Orient. Pour qu’une introgression se produise, deux entités biologiques ayant un ancêtre commun relativement récent (par exemple ici, Homo erectus) doivent se séparer et rester isolées l’une de l’autre pendant suffisamment longtemps pour que leurs pools génétiques puissent devenir distinctement divergents (Figure 1). Ainsi, le patrimoine génétique de la population réceptrice contient des allèles que l’on peut distinguer comme introgressés et non introgressés. Il est maintenant de plus en plus admis qu’une ascendance néandertalienne demeure dans les génomes eurasiens modernes et les a enrichis en allèles (Green et al., 2010 ; Prüfer et al., 2014 ; Vernot et Akey, 2014). Les séquences introgressées ont réintroduit des milliers d’allèles ancestraux qui avaient été perdus dans les populations eurasiennes et en particulier au moment d’une réduction massive de population (Rinker et al., 2019)[18]. Cette perte d’allèles semble s’être produite il y a environ 75 000 ans, un peu avant la seconde migration hors d’Afrique. Seuls 3 000 à 10 000 individus auraient survécu à la suite de changements climatiques majeurs ou peut-être à la suite de l’éruption cataclysmique du Toba en Indonésie (Gagneux et al., 1999 ; Harpending and Rogers, 2000 ; Jorde et al., 1998 ; Quintana-Murci et al., 1999 ; Underhill et al., 2000)[19].
Ces séquences introgressées d’origine néandertalienne codent diverses fonctions qu’il est possible de déterminer par des analyses complexes demandant cependant confirmation. Selon Khrameeva et al. (2014), les Néandertaliens auraient acquis des modifications du catabolisme lipidique bénéfiques pour la survie en climat froid. Les variants alléliques correspondants auraient ensuite été acquis par les humains modernes par introgression en provenance des Néndertaliens et rapidement portés à une fréquence élevée par sélection positive. Autrement dit, les hommes modernes ayant acquis par introgression des allèles de résistance au froid, auraient mieux survécus que ceux qui n’en avaient pas reçu et les auraient transmis à leur descendance. Pour vérifier cette hypothèse, Khrameeva et al. ont recherché des signatures de sélection positive dans les génomes des humains contemporains d’origine européenne, asiatique et africaine. Ils ont effectivement trouvé un excès significatif de sélection positive dans les régions génétiques codant le métabolisme lipidique des Européens contemporains, mais pas des Asiatiques ni des Africains. Simonti et al. (2016) ont étudié 1000 phénotypes provenant des dossiers médicaux d’environ 28 000 adultes d’ascendance européenne. Les allèles néandertaliens expliquent une fraction significative du risque de dépression et de lésions cutanées résultant de l’exposition au soleil (kératose actinique). D’autres allèles néandertaliens sont associés à l’hyper coagulation et au tabagisme.
Par une autre méthode d’analyse et en utilisant un échantillon de plus de 100 000 individus d’ascendance européenne, Dannemann et Kelso (2017) ont montré que des allèles introgressés d’origine néandertalienne contribuaient au déterminisme des caractéristiques de la peau et de celle des cheveux. Ces mêmes auteurs ont aussi trouvé deux allèles archaïques qui contribuent de manière significative aux différences dans le rythme circadien[20]. Ils ont trouvé une corrélation significative entre la fréquence d’un allèle néandertalien et la latitude. Le fait que les populations plus éloignées de l’équateur présentent des fréquences plus élevées de cet allèle néandertalien que les populations proches de l’équateur est en accord avec l’influence de la longueur du jour sur le rythme circadien.
D’autres études génomiques ont aussi démontré que d’autres lignées d’hominidés anciens avaient introgressé avec les hommes modernes. C’est ainsi que des Désinoviens, différents génétiquement des Néandertaliens et identifiés en mars 2010 à partir d’une phalange fossile de 41 000 ans provenant de la grotte de Desinova dans l’Altaï en Sibérie, auraient aussi introgressé avec les hommes modernes[21] 19. Selon Huerta-Sánchez et Gasey (2015), les Tibétains auraient acquis un allèle favorisant la vie en altitude par introgression avec des Désinoviens. D’autre part, Mélanésiens et Aborigènes australiens possèdent des allèles désinoviens, ce qui reste à expliquer. Il est possible que d’autres populations anciennes d’hominidés, encore à découvrir, ont divergé à partir d’Homo erectus, se sont reconnectées, partageant leur matériel génétique les unes avec les autres à maintes reprises par introgression, puis avec les hommes modernes (Figure 3).
Un tel processus, cependant de moindre ampleur, a aussi été récemment mis en évidence dans les populations africaines contemporaines. Selon Hammer et al. (2011) ces populations contiendraient une petite proportion de matériel génétique introgressé il y 35 000 ans à partir d’une population archaïque d’Afrique centrale qui se serait séparée des hommes modernes il y a 700 000 ans (Figure 3). De même, chez les Africains contemporains, les variants du gène MUC7 qui code pour la mucine-7, une des protéines les plus abondantes de la salive, proviendrait d’une introgression en provenance d’une population africaine ancienne inconnue (Xu et al., 2017).
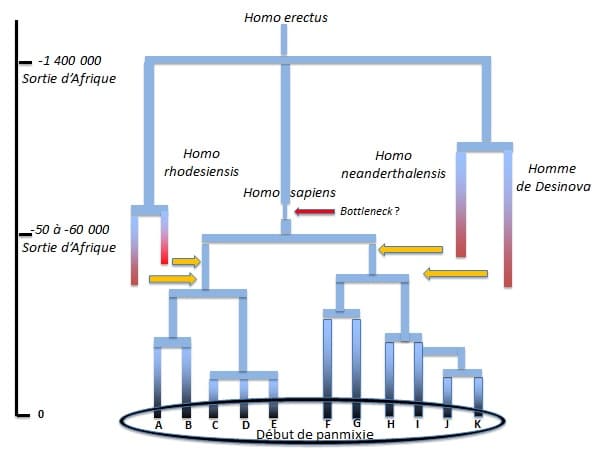
Figure 3. Essai de représentation simplifiée de l’évolution du genre Homo jusqu’à l’époque actuelle.
A : Afrique du sud ; B Afrique de l’Est ; C : Afrique centrale ; D : Afrique centrale ouest ; E : Afrique de l’ouest ; F : Europe ; G : ouest de l’Asie ; H : Asie centrale ; I : Polynésie ; J : Est de l’Asie ; K : Amérique. Les introgressions sont représentées par des flèches jaunes.
Conclusions
L’avènement de la génétique des populations associée à de puissants traitements informatiques et celui de la génomique appliquée à l’ADN moderne et à l’ADN fossile ont révolutionné les connaissances que nous avions de notre histoire, qui pendant deux siècles a été essentiellement basée sur la paléontologie classique.
Les variations génétiques ou plus exactement les variations alléliques dont les Eurasiens actuels sont porteurs proviennent d’une part des populations originelles modernes qui ont quitté l’Afrique il y a environ 50 000 ou 60 000 ans et d’autre part d’au moins deux populations anciennes qui ont échangé des allèles entre elles puis avec les hommes modernes en provenance d’Afrique. Populations modernes venant d’Afrique et populations anciennes eurasiennes ont cependant la même origine à savoir une première sortie d’Afrique il y a environ 1,4 millions d’années par des hommes appartenant à l’entité Homo erectus. Ces Homo erectus ont évolué en Afrique pour aboutir à la naissance sur ce même continent, il y a 150 000 à 200 000 ans, d’Homo sapiens, l’homme moderne.
Parallèlement en Eurasie, l’entité Homo erectus a évolué vers des formes nouvelles dont l’homme de Neandertal apparu il y a environ 450 000 ans, l’homme de Desinova et probablement d’autres entités que nous ne connaissons pas encore. Ces Eurasiens ont rencontré les Africains d’abord au Moyen-Orient, puis un peu partout lorsque ces hommes modernes d’origine africaine ont colonisé l’ensemble de l’Eurasie. Les échanges alléliques par introgression ont été, semble-il, multiples entre modernes et anciens, les anciens enrichissant en allèles les modernes dont la panoplie aurait été réduite il y a 75 000 ans au moment du bottleneck ou goulot d’étranglement qui aurait considérablement réduit la population moderne africaine[22].
Enrichis en allèles, les homme modernes ont colonisé le monde, ce qui a eu pour conséquence l’apparition de nouveaux allèles par isolement géographique. La diversité allélique est devenue considérable avec constitution de populations régionales multiples et séparées les unes des autres.
Notre histoire est loin d’avoir été complètement décryptée. Nous continuons d’acquérir des données sur la structuration allélique du monde et sur les ADN fossiles. A l’heure actuelle, nous manquons néanmoins de données génomiques provenant d’hominidés anciens d’Afrique, d’Asie du Sud-Est et d’Océanie. Nous disposons cependant déjà de données et d’outils permettant de lever une partie du voile recouvrant la complexité sous-estimée de notre passé. Nul doute que de nouvelles découvertes viendront compléter bientôt cette histoire très complexe.
Après cette diversification, probablement unique dans l’histoire du monde animal, cette situation évolue maintenant dans le sens inverse. Les conquêtes, les migrations voulues ou imposées, l’ouverture des frontières et les déplacements facilités à courte ou longue distance, ont pour conséquence un brassage allélique généralisé. Ce brassage tend en effet à homogénéiser les fréquences alléliques entre populations et s’opposent à la différenciation génétique qui se produit entre populations sous l’effet des divers facteurs évolutifs.
Ce brassage est variable suivant les continents ou les pays. Mais là où il a lieu, il a pour conséquence une augmentation de la variabilité allélique et donc du polymorphisme. Autrement dit, au sein des nouvelles entités les individus sont de plus en plus divers. Un nouvel état panmictique s’établira progressivement tout en évoluant en fonction des forces évolutives qui demeureront et en fonction des cultures, des religions de modes de vie ou des changements environnementaux.
Références
Banks, W. E., d’Errico, F., Peterson, A. T., Kageyama, M., Sima, A., & Sánchez-Goñi, M. F. (2008). Neanderthal extinction by competitive exclusion. PLoS One, 3 (12), e3972.
Brunet, M., Guy, F., Boisserie, J. R., Djimdoumalbaye, A., Lehmann, T., Lihoreau, F., … & Zollikofer, C. (2004). Toumaï, Miocène supérieur du Tchad, le nouveau doyen du rameau humain. Comptes Rendus Pale vol, 3 (4), 277-285.
Brunet, M., Guy, F., Pilbeam, D., Mackaye, H. T., Likius, A., Ahounta, D., … & Zollikofer, C. (2002). A new hominid from the Upper Miocene of Chad, Central Africa. Nature, 418 (6894), 145-151.
Dannemann, M., Prüfer, K., & Kelso, J. (2017). Functional implications of Neandertal introgression in modern humans. Genome biology, 18 (1), 1-11.
Duda, P., Jan Zrzavý, Human population history revealed by a supertree approach. Sci Rep 6, 29890 (2016). https://doi.org/10.1038/srep29890.
Gagneux, P., Wills, C., Gerloff, U., Tautz, D., Morin, P. A., Boesch, C., … & Woodruff, D. S. (1999). Mitochondrial sequences show diverse evolutionary histories of African hominoids. Proceedings of the National Academy of Sciences, 96 (9), 5077-5082.
Gokcumen, O. Archaic hominin introgression into modern human genomes. Yearbook Phys Anthropol. 2020;171(Suppl. 70): 60–73. https://doi.org/10.1002/ajpa.23951.
Green, R. E., Krause, J., Ptak, S. E., Briggs, A. W., Ronan, M. T., Simons, J. F., … & Pääbo, S. (2006). Analysis of one million base pairs of Neanderthal DNA. Nature, 444 (7117), 330-336.
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., … & Pääbo, S. (2010). A draft sequence of the Neandertal genome. science, 328 (5979), 710-722.
Hammer, M. F., Woerner, A. E., Mendez, F. L., Watkins, J. C., & Wall, J. D. (2011). Genetic evidence for archaic admixture in Africa. Proceedings of the National Academy of Sciences, 108 (37), 15123-15128.
Harpending, H., & Rogers, A. (2000). Genetic perspectives on human origins and differentiation. Annual review of genomics and human genetics, 1(1), 361-385.
Huerta-Sánchez, E., & Casey, F. P. (2015). Archaic inheritance: supporting high-altitude life in Tibet. Journal of Applied Physiology, 119 (10), 1129-1134.
Johannesson, K. (2001). Parallel speciation: a key to sympatric divergence. Trends in Ecology & Evolution, 16 (3), 148-153.
Jorde, L. B., Bamshad, M., & Rogers, A. R. (1998). Using mitochondrial and nuclear DNA markers to reconstruct human evolution. Bioessays, 20 (2), 126-136.
Khrameeva, E. E., Bozek, K., He, L., Yan, Z., Jiang, X., Wei, Y., … & Khaitovich, P. (2014). Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nature communications, 5 (1), 1-8.
Lumley de, H. (2006). Il y a 400 000 ans : la domestication du feu, un formidable moteur d’hominisation. Comptes Rendus Pale vol, 5 (1-2), 149-154.
McCoy, R. C., Wakefield, J., & Akey, J. M. (2017). Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell, 168 (5), 916-927.
Mayr, E., 1942, Systematics and the Origin of Species. New York: Columbia University Press.
Meyer, M., Kircher, M., Gansauge, M. T., Li, H., Racimo, F., Mallick, S., … & Pääbo, S. (2012). A high-coverage genome sequence from an archaic Denisovan individual. Science, 338 (6104), 222-226.
Plagnol, V., & Wall, J. D. (2006). Possible ancestral structure in human populations. PLoS genetics, 2 (7), e105.
Plomion, C., Aury, J. M., Amselem, J., Leroy, T., Murat, F., Duplessis, S., … & Salse, J. (2018). Oak genome reveals facets of long lifespan. Nature Plants, 4 (7), 440-452.
Prüfer, K., Racimo, F., Patterson, N., Jay, F., Sankararaman, S., Sawyer, S., … & Pääbo, S. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature, 505 (7481), 43-49
Quintana-Murci, L., Semino, O., Bandelt, H. J., Passarino, G., McElreavey, K., & Santachiara-Benerecetti, A. S. (1999). Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nature genetics, 23 (4), 437-441.
Rinker, D. C., Simonti, C. N., McArthur, E., Shaw, D., Hodges, E., & Capra, J. A. (2019). Neanderthal introgression reintroduced functional alleles lost in the human out of Africa bottleneck. bioRxiv, 533257.
Senut, B., Pickford, M., Gommery, D., Mein, P., Cheboi, K., & Coppens, Y. (2001). First hominid from the Miocene (Lukeino formation, Kenya). Comptes Rendus de l’Académie des Sciences-Series IIA-Earth and Planetary Science, 332 (2), 137-144.
Simonti, C. N., Vernot, B., Bastarache, L., Bottinger, E., Carrell, D. S., Chisholm, R. L., … & Capra, J. A. (2016). The phenotypic legacy of admixture between modern humans and Neandertals. Science, 351 (6274), 737-741.
Spoor, F., Leakey, M. G., Gathogo, P. N., Brown, F. H., Antón, S. C., McDougall, I., … & Leakey, L. N. (2007). Implications of new early Homo fossils from Ileret, east of Lake Turkana, Kenya. Nature, 448 (7154), 688-691.
Taskent, R. O., & Gokcumen, O. (2017). The multiple histories of WesternAsia: Perspectives from ancient and modern genomes.Human Biology, 89, 107–117.
Tattersall, I. (2009). Human origins: Out of Africa. Proceedings of the National Academy of Sciences of the United States of America,106,16018–16021.
Tattersall, I., & Schwartz, J. H. (1999). Hominids and hybrids: The place of Neanderthals in human evolution. Proceedings of the National Academy of Sciences of the United States of America, 96, 7117–7119.
Templeton, A. (2002). Out of Africa again and again. Nature, 416,45–51.
Turelli, M., Barton, N. H., & Coyne, J. A. (2001). Theory and speciation. Trends in ecology & evolution, 16 (7), 330-343.
Underhill, P. A., Shen, P., Lin, A. A., Jin, L., Passarino, G., Yang, W. H., … & Oefner, P. J. (2000). Y chromosome sequence variation and the history of human populations. Nature genetics, 26 (3), 358-361.
Veeramah, K. R., & Hammer, M. F. (2014). The impact of whole-genome sequencing on the reconstruction of human population history. Nature Reviews. Genetics,15, 149–162.
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., … & Kalush, F. (2001). The sequence of the human genome. Science, 291 (5507), 1304-1351.
Venter, J. C., Smith, H. O., & Adams, M. D. (2015). The sequence of the human genome. Clinical chemistry, 61 (9), 1207-1208.
Vernot, B., & Akey, J. M. (2014). Resurrecting surviving Neandertal lineages from modern human genomes. Science, 343, 1017–1021.
Vernot, B., Tucci, S., Kelso, J., Schraiber, J. G., Wolf, A. B., Gittelman, R. M. ,… Akey, J. M. (2016). Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science, 3 52, 235–239.
Villanea, F. A., & Schraiber, J. G. (2019). Multiple episodes of interbreeding between Neanderthal and modern humans. Nature Ecology & Evolution, 3,39–44.
Wall, J. D., & Yoshihara Caldeira Brandt, D. (2016). Archaic admixture inhuman history. Current Opinion in Genetics & Development, 41,93–97.
Willerslev, E., & Cooper, A. (2005). Review Paper. Ancient DNA. Proceedings of the Royal Society B: Biological Sciences, 272, 3–16.
Wolf, A. B., & Akey, J. M. (2018). Outstanding questions in the study of archaic hominin admixture. PLoS Genetics,14, e1007349.
Wolpoff, M. H., Hawks, J., & Caspari, R. (2000). Multiregional, not multiple origins. American Journal of Physical Anthropology, 112, 129–136.
Xu, D., Pavlidis, P., Taskent, R. O., Alachiotis, N., Flanagan, C., DeGiorgio, M ,… Gokcumen, O. (2017). Archaic Hominin Introgression in Africa contributes to functional salivary MUC7 genetic variation.Molecular Biology and Evolution, 34, 2704–2715.
Xing, J., Watkins, W. S., Shlien, A., Walker, E., Huff, C. D., Witherspoon, D. J., … & Jorde, L. B. (2010). Toward a more uniform sampling of human genetic diversity: a survey of worldwide populations by high-density genotyping. Genomics, 96 (4), 199-210.
[1] Huit volumes supplémentaires ont été publiés à titre posthume.
[2] On a longtemps pensé qu’Homo erectus était un descendant d’Homo habilis. Les découvertes faites au Kenya près du lac Turkana démontrent qu’Homo habilis et Homo erectus ont cohabité en Afrique pendant 500 000 ans (Spoor et al., 2007, Nature, 448, 688-691. Homo ergaster est souvent confondu avec Homo erectus. Il pourrait l’avoir précédé. L’entité Homo erectus ne fait pas l’unanimité. Mais la tendance est de regrouper sous ce vocable toutes les formes du genre Homo apparues il y a environ 2 millions d’années ou plus et qui ont commencé à se disséminer en Europe et en Asie il y a environ un million d’années. L’entité Homo erectus regroupe ce qui avait été dénommé Pithécanthrope ou Sinanthrope.
[3] La dénomination Homo heidelbergensis ne fait pas consensus et l’homme de Heidelberg semble bien appartenir à l’entité Homo erectus.
[4] L’homme de Tautavel, souvent considéré comme appartenant à Homo heidelbergensis, fait aussi partie de l’entité Homo erectus.
[5] Henry de Lumley (2006). Il y a 400 000 ans : la domestication du feu, un formidable moteur d’hominisation, Comptes Rendus de Paléontologie, vol, 5, 1-2, 149-154.
[6] Chez l’homme, les individus sont diploïdes, c’est-à-dire possède à chaque locus deux allèles ou variants d’un même gène. Chez les mousses ou certains champignons, les individus sont haploïdes et ne possèdent qu’un allèle à un locus.
[7] Chez les arbres âgés, il peut y avoir modification du génome de certaines cellules somatiques (Plomion et al., 2018).
[8] Mais en général on caractérise le génotype d’un individu par une toute petite partie de son information génétique et le plus souvent par quelques dizaines de séquences courtes appelées microsatellites (séquences de 2 à 6 nucléotides répétées).
[9] Un allèle est dominant lorsque le caractère qu’il code s’exprime même s’il n’est présent que sur un seul allèle du locus. Un allèle est récessif lorsqu’il ne s’exprime que s’il est présent sur les deux allèles du locus.
[10] État d’une population où les individus sont répartis de manière homogène et se reproduisent aléatoirement.
[11] Il est possible d’établir l’ascendance d’un individu, d’une population ou d’une espèce par analyse phylogénétique en utilisant quelques séquences variables du génome comme celles qui codent l’ADN ribosomal. Il est aussi possible d’utiliser le génome entier lorsqu’il est disponible.
[12] Le génome humain comprend 2,85 Gbp (milliard de paires de base) et environ 26 000 gènes codant pour des protéines.
[13] L’estimation du nombre de gènes chez l’homme n’est pas définitive. Actuellement cette estimation est comprise entre 20 et 21 000 gènes.
[14] Le Single Nucleotide Polymorphism ou SNP ou polymorphisme d’un seul nucléotide (PSN) est la variation sur un locus d’une seule paire de bases entre individus d’une même population ou entité biologique. Chez l’homme, il existe un SNP toutes les mille paires de bases.
[15] La diversification des populations africaines subsahariennes est loin d’avoir été déchiffrée et apparaît très complexe.
[16] Les génomes eurasiens pourraient contenir une centaine de milliers d’allèles néanderthaliens.
[17] L’introgression ou introgressive hybridization est le transfert d’allèles d’une espèce à une autre non encore complètement génétiquement séparée, ce qui rend l’inter fécondation possible. Une première hybridation est suivie de rétrocroisements avec des individus de l’espèce hôte, ces derniers restant majoritaires.
[18] McCoy, Wakefield et Akey (2017) ont identifiés dans 767 gènes 1236 SNPs (Single Nucleotide Polymorphism) introgressés soit 24,5 %.
[19] L’existence même d’un goulot d’étranglement résultant d’une réduction drastique de population ne fait pas l’unanimité.
[20] Le rythme circadien est un cycle régissant le sommeil et l’alimentation.
[21] Une couverture quasi complète du génome désinovien a été publiée en 2012 par Meyer et al. L’homme de Harbin, Homo longi, découvert en Chine et publié en 2012, pourrait être apparenté aux Désinoviens.
[22] L’existence de ce goulot d’étranglement ne fait pas consensus. Sa date est aussi controversée. Ce bottleneck aurait eu lieu il y a environ 75 00 ans avant la seconde sortie d’Afrique ou il y a 60 000 ans au moment de cette seconde sortie d’Afrique.